



For Researchers


Despite its ultrarare prevalence, HSPB8 myopathy is an area of active research. Several academic groups in the USA and Europe work on elucidating the mechanisms behind the disease and developing treatments. The results of their work are published in scientific journals, below we list key academic groups and publications.
One treatment avenue researchers are pursuing is using trehalose to ameliorate the symptoms. Here you can find our summary about trehalose and HSPB8 myopathy.
Introduction
HSPB8 Myopathy (Heat Shock Protein family B (small) member 8), or Myofibrillar Myopathy type 13 with Rimmed Vacoles (MFM13) is an ultra-rare, adult-onset, muscle wasting condition caused by mutations in HSPB8 gene. Inherited in autosomal dominant fashion, HSPB8 Myopathy was first described in 2016 by Ghaoui et al., and since then eight case studies have been published, describing around 30 patients worldwide (Ghaoui et al, 2016; Echaniz-Laguna et al, 2017; Cortese et al, 2018; Al-Tahan et al, 2019; Nicolau et al, 2020; Inoue-Shibui et al, 2021, Tan et al., 2024, Yang etal., 2024 ). However, HSPB8 Myopathy is not routinely included in genetic myopathy panels and therefore heavily underdiagnosed.
Clinical Presentation
HSPB8 Myopathy typically manifests in the second, third, or fourth decade of life. It is characterized by progressive muscle weakness, which may initially appear either proximally or distally. The condition predominantly affects the lower limbs and gradually spreads to the truncal muscles and upper limbs. Due to its progressive nature, individuals experience increasing difficulty with muscle strength and mobility over time. More clinical information can be found in the ‘for clinicians’ section.
Histological Findings
Histological examination of HSPB8 Myopathy reveals features characteristic of myofibrillar myopathy. Muscle biopsies typically show fatty replacement, aggregates, rimmed vacuoles, and endomysial fibrosis (Al-Tahan et al, 2019). The pathology observed in HSPB8 Myopathy is similar to that seen in myopathies caused by gene defects in other components of the CASA complex, such as BAG3 and DNAJB6.
HSPB8 Protein and the CASA Complex


HSPB8 protein belongs to the family of HSPBs, consisting of ten members. HSPBs recognize misfolded proteins in near-native state, prevent their future misfolding and facilitate refolding by HSPA/HSP70 (Tedesco et al, 2023b).
HSPB8 forms a stable complex, knows as the Chaperone Assisted Selective Autophagy (CASA) complex with chaperone HSPA, cochaperone BAG3 and E3 ubiquitin ligase STUB1. CASA is a highly selective pathway for disposal of misfolding and aggregating proteins, mostly studies in the skeletal muscle and neurons.


Figure 1 - From Tedesco et al., 2023b The chaperone-assisted selective autophagy (CASA) complex. (A) Schematic representation of the CASA complex. BAG3 (green) mediates the assembly of the CASA complex, thereby linking chaperones of the HSPA family (purple) to small heat shock proteins such as HSPB8 (blue). The HSPA-associated E3 ubiquitin ligase STUB1 (orange) interacts with the complex for the ubiquitination of the bound client protein. (B) Engulfment of the CASA complex by LC3-decorated phagophore membrane is facilitated by the autophagic cargo receptor SQSTM1, leading to formation of double-membrane vesicles (called autophagosomes) and final degradation of its content by fusion with the lysosomes
In muscle, CASA maintains muscle integrity by facilitating turnover of structural muscle components damaged by mechanical strain. In neurons, it promotes the degradation of aggregating substrates that cause neurodegenerative diseases.
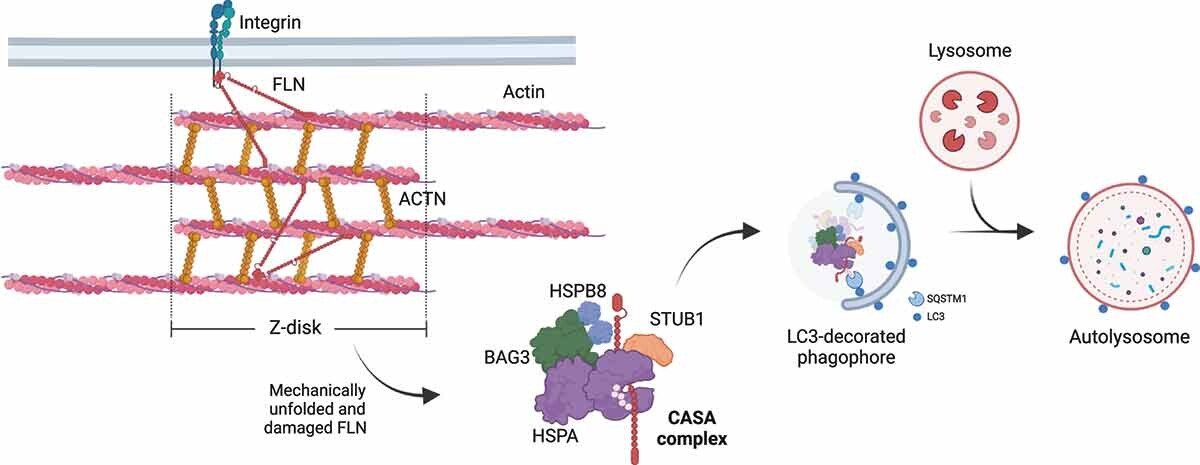
Figure 2-From Tedesco et al., 2023 - CASA-mediated degradation of Z-disk component FLNC. Mechanically unfolded and damaged forms of FLN are recognized and degraded by the CASA pathway. Here, BAG3 closely cooperates with HSPB8 (the “holdase”) and regulates the ATP-dependent chaperone cycle of HSPA, necessary for substrate processing. STUB1-mediated ubiquitination and subsequent recruitment of the autophagic receptor SQSTM1 initiates autophagosome formation, followed by lysosomal fusion for degradation.
Damaged or misfolded proteins are recognized by HSPB8 and HSPA and can be subjected to refolding (driven by HSPA) or ubiquitination (driven by STUB1). Ubiquitinated substrates are degraded via the autophagosome-lysosomal pathway. In case of degradation failure, the misfolded substrates are compartmentalized into aggresomes. The CASA complex is also involved in stress granules maintenance (granulostasis) (Tedesco et al, 2023b).
Mutations in the CASA complex have been linked to several human diseases. Mutations in BAG3 lead to myopathies, neuropathies and cardiomyopathies (reviewed in Tedesco et al, 2023b). Similarly, mutations in HSPB8 lead to neuropathies, and myopathies. However, mutations in STUB1 result in a different phenotype – spinocerebellar ataxia. CASA also is being investigated as a pharmacological target. Several compounds have been found to upregulate HSPB8 expression, including colchicine, doxorubicin and trehalose (Crippa et al, 2016; Chierichetti et al, 2023).
Mechanism of HSPB8 Myopathy Pathology
Mutations in HSPB8 gene have been previously linked to a rare type of Charcot Marie Tooth disease (CMT type 2L) and distal hereditary autosomal dominant neuronopathy type 2 (OMIM). HSPB8 Myopathy is caused by mutations in HSPB8 gene, typically frameshift mutations in exon 3 (Figure 1). This mutation leads to a stop codon readthrough and results in a longer protein product, with extended C terminal region, that is predicted to be highly disordered and more prone to aggregation than the WT protein (Tedesco et al, 2023a).
List of mutations known to cause HSPB8 Myopathy (personal communication with dr Lan Weiss, 2024):
c.515dupC (p.P173Sfs*43)
c.151insC (p.P173SfsX43)
c.508_509delCA (p.GIn170Glyfs*45)
c.525-529delAACAT (p.T176Wfs*38)
c.421A>G (p.K141E)
c.515delC (p.Pro172Leufs*75)
c.577-580dupGTCA (p.T194Sfs*23)


Figure From Inoue-Shibui et al, 2021- HSPB8 variants and predicted proteins. A Scheme of HSPB8, HSPB8, and sHSPs. HSPB8 consists of three exons. The previously reported muscular phenotype related to c.515dup (p.Pro173Serfs*43), c.508_509del (p.Gln170Glyfs*45), and c.577_580dup (p.Thr194Serfs*23) are located in the last exon, as a result of frameshift mutations. Hot spot mutations of Charcot-Marie-Tooth type 2L and distal hereditary motor neuropathy IIa are located at the 421 and 423 bases in exon 2. HSPB8 contains an α-crystallin domain, in grey. The dark blue parts in the α-crystallin domain are β4and β8 chains, respectively, to which the IXI/V domain binds. sHSPs, other than HSPB8, have an α-crystallin domain and an IXI/V domain (yellow part) in the Cterminal. B Predicted amino acid sequences. The reference sequence (CCDS9189.1) consists of 196 amino acids. The amino acids of previously reported variants are in red. The variant identified in this study, c.525_529del (p.Thr176Trpfs*38) is located in the latter part of the last exon causing a frameshift mutation, and is predicted to result in the elongation of 17 amino acids in the C-terminal. The previously reported p.Gln170Glyfs*45, p.Pro173Serfs*43, and p. Thr194Serfs*23 are also predicted to have an elongation effect on amino acids in the C-terminus. The IXI/V motifs are boxed yellow.
Mechanisms of HSPB8 Myopathy pathology are under investigation. In 2017, Echaniz-Laguna suggested that myopathy is caused by haploinsufficiency of HSPB8 protein, as they observed decreased levels of HSPB8 protein in patient muscle tissue compared to controls, and were not able to detected a shorter or a longer form using an antibody targeting the N-terminal of HSPB8 (Echaniz-Laguna et al, 2017). However a recent study by Tedesco and colleagues points to a dominant negative and toxic gain of function mechanism (Tedesco et al, 2023a).
This study investigated the biochemical and functional alterations associated with the HSPB8_fs mutant proteins. It shows that HSPB8_fs mutants are highly insoluble and tend to form proteinaceous aggregates in the cytoplasm. Although all HSPB8 frameshift mutants retain their ability to interact with CASA members, they sequester these members into HSPB8-positive aggregates along with two autophagy receptors, SQSTM1/p62 and TAX1BP1. This process adversely affects CASA’s ability to remove its clients, leading to a general failure in proteostasis response. The aggregation of these mutants is an intrinsic feature of the mutated amino acid sequence and occurs independently of interactions with other CASA members or autophagy receptors (Tedesco et al, 2023a).

Figure 4 From Tedesco et al., 2023a - Frameshift mutants of HSPB8 form high molecular weight insoluble species and cytoplasmic aggregates. (A) Schematic representation of HSPB8_WT and its HSPB8_fs mutant structures. The α-crystallin domain (ACD) is reported in Orange, the N-terminal region (NTR) in blue, the C-terminal region (CTR) in green. The striped green region represents the mutated CTR of p.P173Sfs*43 (fs1), p.Q170Gfs*45 (fs3) and p.T176Wfs*38 mutants. The red region represents the common C-terminal extension (CE) shared by all fs mutants. The box at the bottom reports the nomenclature of the mutated C-termini of the HSPB8 mutants: mCTR (mutated CTR, comprises both the mutated CTR and the CE) and the CE. (B) Immunofluorescence analysis of HeLa cells transiently transfected with V5-tagged HSPB8 constructs. HSPB8 is in green, nuclei were stained with DAPI, scale bar: 20 μm. (C) Western blot and filter retardation assay (FRA) analyses of NP-40 soluble/insoluble protein fractions of HeLa cells transiently transfected with V5-tagged HSPB8 constructs or an empty vector (EV). Bar graphs report mean values (± SD) of densitometry of HSPB8-V5 on soluble TUBA/tubulin alpha for western blot. All graphs are normalized to the HSPB8_WT-V5. One-way ANOVA with Tukey’s test was performed: * p < 0.05, ** p < 0.01, *** p < 0.001; n = 3.
Schematic representation of mechanism of HSPB8 Myopathy pathology

Research Toolkit
For researchers interested in studying HSPB8 Myopathy, below is the list of resources currently available. While we are doing our best to make sure information in this table is up to date, please feel free to reach out to us for me most recent information.

Cells and DNA available from Coriell Cell Repository

Mice
Knock out mouse model: C57BL/6NJ-Hspb8em1(IMPC)J/Mmjax (Jackson Lab): MMRRC Strain #051194-JAX, RRID: MMRRC_051194-JAX- (Bouhy et al, 2018)
Key characteristics of the mice:
Lack of motor or sensory phenotype or CMAP abnormalities, normal behaviour and physiology. No myopathic or neuropathic phenotype
absence of atrophic or necrotic fibres in garstrocnemius via EM analysis
accumulation of pathologic mitochondria many of which presented with degenerating cristae and abnormal matrix in gastrocnemius
an increased susceptibility to heart failure under the specific context of cardiac overload (Qiu et al., 2011)
HSPB8 Myopathy Library
Case studies
1) Neurology: Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy
Ghaoui et al, 2016
2) Neuropathologica: HSPB8 haploinsufficiency causes dominant adult-onset axial and distal myopathy
Echaniz-Laguna et al, 2017
Cortese et al, 2018
4) Neurology genetics: New family with HSPB8-associated autosomal dominant rimmed vacuolar myopathy
Al-Tahan et al, 2019
Nicolau et al., 2020
6) A novel deletion in the C-terminal region of HSPB8 in a family with rimmed vacuolar myopathy
Inoue-Shibui et al., 2021
7) A novel c.515delC HSPB8-multisystem proteinopathy associated with inclusion body myopathy with cardiomyopathy Tan et al., 2024
Molecular pathology
Tedesco et al, 2023
Reviews
1) The chaperone-assisted selective autophagy complex dynamics and dysfunctions Tedesco et al., 2023, Autophagy
Key Academic Groups

-
Dr. Virginia E. Kimonis:
Dr. Kimonis received her medical degree from Southampton Medical School in 1976, completed her training in pediatrics and general practice at leading institutions in the UK. In the US she completed training in advanced pediatrics at Massachusetts General Hospital/ Harvard Medical School. Her fellowship in biochemical and clinical genetics was at the National Institute of Health, John’s Hopkins and National Children’s Hospital. She was the Division Chief of Genetics and Genomic Medicine at SIU School of Medicine, Springfield, IL and at UCI School of Medicine, Irvine, CA. Her research interests encompass rare genetic neuromuscular disorders, in addition to other rare diseases.
Kimonis Lab: The Kimonis Laboratory, part of the Division of Genetics & Genomic Medicine at UCI School of Medicine, investigates genetic causes and treatments for rare muscle diseases such as rimmed vacuolar myopathy caused by HSPB8 variants, and inclusion body myopathy caused by VCP variants and other genetic disorders.
The lab's clinical research spans genetic causes, natural history, and treatments for rare diseases, including hereditary myopathies, lysosomal storage diseases, and Prader-Willi Syndrome, and other rare genetic disorders.
Since discovering the link between Inclusion Body Myopathy and HSPB8 gene mutations in the first family, the lab generated CRISPR mouse models, induced pluripotent stem cell (iPSCs) to generate myoblasts or muscle cells. These important tools are used for developing novel treatments like colchicine, arimoclomol, and gene targeting technology for HSPB8 myopathy.
Alyaa Shmara:
Alyaa is a postdoctoral fellow at the laboratory of Dr. Virginia Kimonis at UCI. Her research focus is studying the mechanisms underlying hereditary inclusion body myopathies associated with Valosin Containing Protein (VCP) and Heat Shock Protein B8 (HSPB8) mutations. She is heavily involved in investigating therapeutic approaches using FDA approved medications, small molecules, and gene therapy with potential to ameliorate pathology in myopathy mouse models.
She also conducts clinical research in Lysosomal Storage Diseases and has completed a Lysosomal Storage Diseases fellowship at UCI’sDepartment of Pediatrics, Division of Genetics and Genomic Medicine from 11/2021-12/2022.
She serves as a patient advocate for Cure HSPB8 myopathy and leads the effort of raising awareness and increasing patient identification of this rare disease.
Alyaa Shmara (researchgate.net)
Pallabi Pal, Ph.D., Project Scientist
Research Interest: Therapeutic strategies of genetic neuromuscular disorders
The underlying mechanisms and the treatment for HSPB8 associated rimmed vacuolar myopathies associated with muscle atrophy and early demise remain to be elucidated. Her goal is to develop a potent therapy to stop/reduce the progression of rimmed vacuolar myopathy. To investigate in vitro the molecular mechanism of HSPB8-associated myopathy and to assess the potential of new treatments, patient iPSC-derived myoblasts are utilized. She is recapitulating the pathogenic phenotype of patient-derived iPSCs differentiated into muscle cells. Compounds that stimulate autophagy favor the removal of protein aggregates. She is currently using an autophagy modifier to facilitate removal of protein aggregates by stimulating the autophagy pathway. Successful completion of the present mechanistic and translational study will pave the way for the treatment of HSPB8-associated inclusion body myopathy and will also benefit other related disorders.
Dr. Lan Weiss
A scientist currently affiliated with the University of California, Irvine. Holding a Doctor of Medicine degree from Hue University, Vietnam, and a Doctor of Philosophy in Human Genetics from Nagasaki University, Japan, Dr. Weiss brings a wealth of knowledge to her current role. Since joining the Kimonis lab in 2016, Dr. Weiss has dedicated her research efforts to unraveling the pathological mechanisms underlying hereditary Rimmed Vacuolar Myopathy associated with mutations in the Heat Shock Protein B8 (HSPB8) gene. Her primary objective is to make significant contributions to the development of a cure for this debilitating disease.
Dr. Weiss is particularly enthusiastic about harnessing the potential of Adeno-Associated Viruses (AAV), renowned for their proven safety and efficacy, to deliver therapeutic genes specifically to the affected muscles—the primary organ impacted in many HSPB8 patients. Additionally, she actively explores the realm of cell therapy, which involves the engraftment of healthy cells to restore or improve muscle function. Her research unfolds across two pivotal pre-clinical platforms: Patient-induced pluripotent stem cell (iPSC)-derived skeletal muscle progenitor cells and the CRISPR Cas knock-in Hspb8 mouse model. This multidimensional approach reflects her dedication to advancing the comprehension and treatment of HSPB8-related conditions.
-
Angelo Poletti, PhD
Professor Angelo Poletti is a Full Professor of Experimental Biology at the University of Milan, specializing in the role of the protein quality control system in neurodegenerative and neuromuscular diseases. He obtained his Italian Laurea and subsequent degrees in Chemical and Pharmaceutical Technology and Experimental Endocrinology, including a Ph.D. in Endocrinological Sciences. With a background that includes three years of research at Baylor College of Medicine in Houston, TX, USA, Professor Poletti now directs the research laboratory of Experimental Biology at the Dipartimento di Scienze Farmacologiche e Biomolecolari and the Centre of Excellence on Neurodegenerative Diseases at the University of Milan, Italy. At the same University, he also teaches 'Experimental Biology' and 'Cell Biology' to students of Biotechnology and Pharmaceutical Chemistry.
Dr. Poletti's research group focuses on several key projects. They investigate motoneuronal degeneration in Spinal and Bulbar Muscular Atrophy (SBMA or Kennedy's disease), exploring neurotoxic effects induced by mutations in the androgen receptor. Additionally, they study motoneuronal degeneration in Amyotrophic Lateral Sclerosis (ALS), particularly the impact of mutations in the Superoxide dismutase (SOD1) and in the C9ORF72 genes, as well as the alterations related to the aberrant TDP-43 protein behavior. More recently the group also became interested in neuromuscular diseases and myopathies caused by mutations in the genes encoding for a small heat shock protein (HSPB8) and its co-chaperone BAG3, proteins involved in a peculiar form of autophagy, named chaperone-assisted selective autophagy (CASA). His lab recently identified a protective mechanism of a small heat shock protein (HSPB8) mediated by autophagy, which efficiently clears misfolded protein from cells, broadening our understanding of neurodegenerative diseases. Poletti’s group also delves into the androgenic and estrogenic control of prostate and breast cancer cell growth, examining the mechanisms of action of androgen and estrogen receptors. Moreover, they explore the effects of gonadal steroids on motoneuronal functions, characterizing receptors and steroid-inducible genes in cultured motoneuronal cells.
Barbara Tedesco, PhD
Barbara Tedesco is an Assistant Professor of Experimental Biology at the Dipartimento di Scienze Farmacologiche e Biomolecolari and the Centre of Excellence on Neurodegenerative Diseases at the University of Milan. Her research focuses on the role of proteostasis imbalance in diseases affecting the neuromuscular system.
She obtained her bachelor's and master’s degrees in Pharmaceutical Biotechnology and Drug Biotechnology, respectively. Then, she obtained a PhD in Integrative Biomedical Research.
She joined the laboratory of Professor Angelo Poletti as an undergraduate student, and she completed my predoctoral and doctoral studies by investigating the mechanisms at the basis of motoneuron diseases, neuropathies, and myopathies.
Her predoctoral and doctoral studies have been primarily devoted to investigating the molecular behavior of mutants and variants of the co-chaperone BAG3 and the small heat shock protein HSPB8, which are associated with neuropathies and (cardio)myopathies.
After the PhD degree, she obtained a fellowship from the IRCCS Istituto Neurologico Carlo Besta (Milan), to investigate mutations affecting the neuro-specific kinesin KIF5A gene, which have been identified in different neurologic and neurodegenerative disorders.
Her current studies are also dedicated to investigating the pathogenic mechanisms and therapeutic strategies in spinal and bulbar muscular atrophy and amyotrophic lateral sclerosis, with a particular interest in the molecular mechanisms regulating protein translation machinery.
-
Dr. Michio Hirano currently serves as Chief of the Neuromuscular Division at Columbia University Medical Center's Neurology Department, Co-Director of the CUMC Muscular Dystrophy Association clinic, and Director of the H. Houston Merritt Center for Muscular Dystrophy and Related Diseases.
In addition to his clinical responsibilities, Dr. Hirano's research primarily centers on mitochondrial diseases and genetic myopathies. Regarding HSPB8 myopathy, Dr. Hirano and his colleagues are working with cultured skin fibroblasts to understand chaperone-assisted selective autophagy (CASA) in pathogenic cells compared to controls. They have profiled the expression of autophagy-related proteins in the cultured cells and are currently conducting drug screening studies to identify whether candidate drugs are predicted to ameliorate or worsen the aberrant autophagy in the HSBP8-mutant cells.