




Disease Classification

Orphanet, a database of rare diseases, and National Organization for Rare Diseases (NORD) classify HSPB8 Myopathy as a subtype of Myofibrillar Myopathy (MFM). Some sources describe it as a form of LGMD. Below, Dr. Alyaa Shmara discusses why we believe HSPB8 Myopathy falls under these categories:
HSPB8 Myopathy is also known as
HSPB8 Inclusion Body Myopathy with Rimmed Vacuoles
HSPB8 Rimmed Vacuolar Myofibrillar Myopathy (Ghaoui et al., 2016)
Autosomal dominant distal axonal motor neuropathy-myofibrillar myopathy syndrome (Orphanet)
Limb-girdle rimmed vacuolar myopathy (Nicolau et al., 2020)
Multisystem proteinopathy (MSP) or MSP-like disorder (Chompoopong et al., 2024)
Autosomal dominant rimmed vacuolar myopathy (Al Tahan et al., 2019)
ORPHA:476093
HSPB8 as MFM

Myofibrillar myopathies are a group of rare genetic neuromuscular disorders that may be diagnosed in childhood but most often appear after 40 years of age. These conditions are highly variable but are characterized by a slowly progressive muscle weakness that can involve skeletal muscle (muscles that function to move bones) and smooth muscle (muscle often associated with organs, such as the digestive tract). Skeletal muscle weakness can be present in the limb muscles close to the center of the body (proximal) as well as the muscle farther from the center of the body (distal). A weakening of the heart muscle (cardiomyopathy) is common and may result in an irregular heartbeat (arrhythmia or conduction defects) or congestive heart failure.
HSPB8 is part of the chaperone-assisted selective autophagy (CASA) complex, a vital part of the cellular protein quality control system in mechanically strained cells and tissues such as skeletal muscle, heart, and lung. In muscle, CASA maintains the Z-disk and protein turnover. CASA mediates degradation of the actin cross-linking protein filamin. If mechanical tension results in permanent unfolding of filamin, it is recognized by the CASA chaperone complex and leads to autophagic degradation of damaged proteins [1].
Mutations in HSPB8 gene, which have previously been associated with Charcot-Marie-Tooth type 2L and distal hereditary motor neuronopathy type IIa, have also been reported to associate with autosomal dominant Rimmed Vacuolar Myopathy. Affected patients have distal and proximal limb girdle myopathy. Muscle biopsy displays histologic features of myofibrillar myopathy with aggregates and rimmed vacuoles.
Ghaoui et al identified 2 families with distal weakness progressing to involve proximal muscles with mutations in HSPB8 causing a dual involvement of a peripheral motor neuropathy and a rimmed vacuolar myofibrillar myopathy. Muscle biopsy shows evidence of a myofibrillar autophagic myopathy, rimmed vacuoles, and protein aggregates positive for proteins associated with myofibrillar myopathy such as desmin, myotilin, and α-B-crystallin. These aggregates also contain HSPB8 and other CASA complex partners DNAJB6 and BAG3 [3,4] (Fig 1.) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4776089/


Laguna et al. identified three families with a HSPB8 myopathy. Muscle biopsies showed dystrophic features with atrophy, necrosis and regeneration, internal nuclei, fibrosis, fiber splitting and rimmed vacuoles. No neurogenic aspect was seen. Protein aggregations were revealed using ubiquitin and desmin antibodies (Fig.2: 4–5). TDP-43 antibody revealed coarse cytoplasmic aggregation and linear sarcolemmal expression (Fig.2: 6). EM studies showed focal disruption of the myofibrillar network, Z lines streaming and areas lacking myofibrils containing granulofilamentous material (Fig.2: 7–8). HSPB8 haploinsufficiency causes dominant adult-onset axial and distal myopathy | Acta Neuropathologica (springer.com)
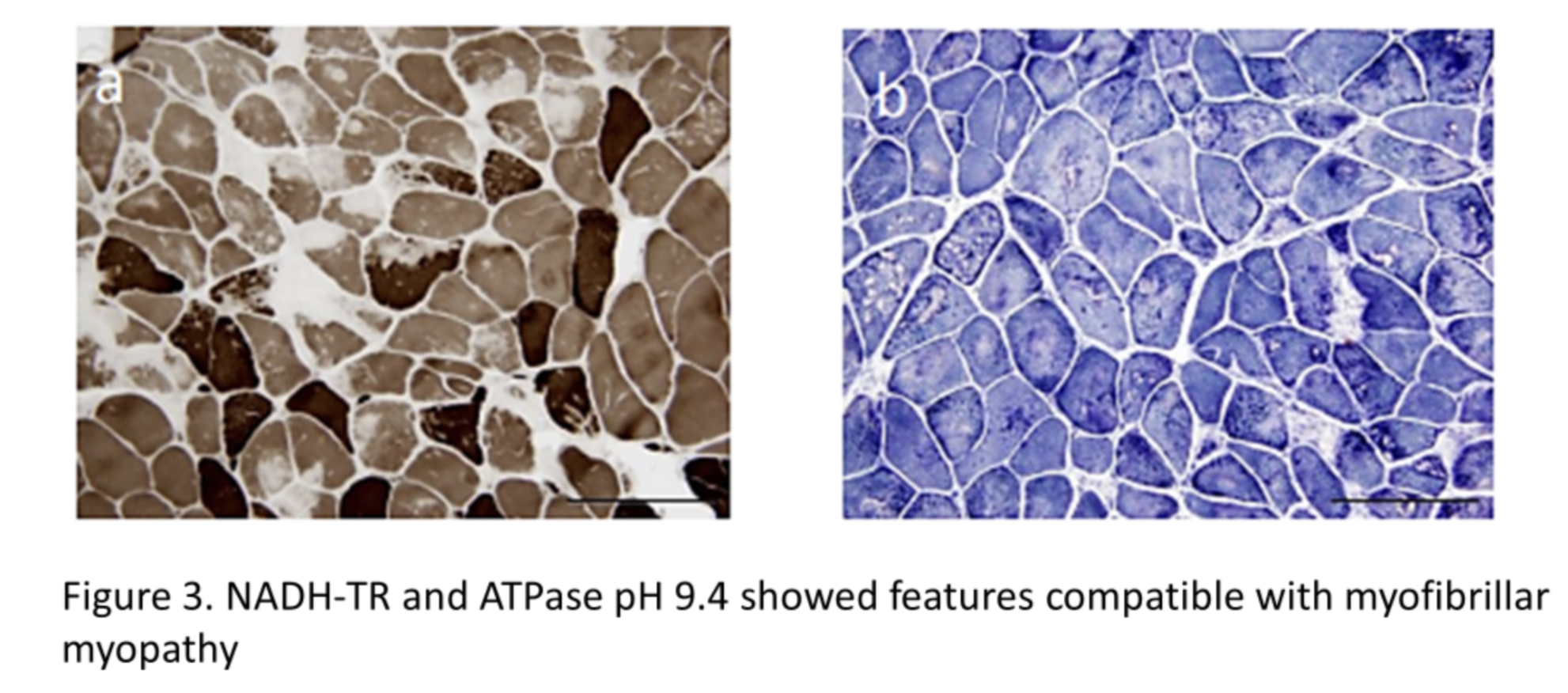
NADH-TR and ATPase pH 9.4 showed features compatible with myofibrillar myopathy [4] (Fig. 3) 401_2017_1724_MOESM1_ESM.pdf (springer.com)
References:
Arndt V, Dick N, Tawo R, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20(2):143-148. doi:10.1016/j.cub.2009.11.022
Sarparanta J, Jonson PH, Golzio C, et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nature Genetics. 2012 Feb;44(4):450-5, S1-2. DOI: 10.1038/ng.1103. PMID: 22366786; PMCID: PMC3315599.
Ghaoui, Roula et al. “Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy.” Neurology vol. 86,4 (2016): 391-8. doi:10.1212/WNL.0000000000002324
Echaniz-Laguna, A., Lornage, X., Lannes, B. et al. HSPB8 haploinsufficiency causes dominant adult-onset axial and distal myopathy. Acta Neuropathol 134, 163–165 (2017). https://doi.org/10.1007/s00401-017-1724-8
Section Author : Dr Alyaa Shmara