





For Clinicians
HSPB8 Myopathy, or Myofibrillar Myopathy Type 13 with Rimmed Vacuoles is an ultra-rare, slowly progressing autosomal dominanant. Mutations in HSPB8 gene, which have previously been associated with Charcot-Marie-Tooth type 2L and distal hereditary motor neuronopathy type IIa, have more recently also been reported to cause Myopathy with Rimmed Vacuoles. Affected patients have distal and proximal limb girdle myopathy. Muscle biopsy displays histologic features of myofibrillar myopathy with aggregates and rimmed vacuoles.
HSPB8 protein is a chaperone involved in the Chaperone Assisted Selective Autophagy (CASA) complex. HSPB8 in conjunction with BAG3 recognizes and promotes the autophagy-mediated removal of misfolded proteins associated with motor neuron disease, Alzheimer, Parkinson, Huntington, and spinocerebellar Ataxia 3.
Step by Step Flowchart


When to Suspect HSPB8 Myopathy?


Clinical characteristics:
Difficulty walking in early adulthood.
Muscle weakness affecting distal lower extremities.
Bilateral foot drop.
Foot and ankle pain.
Difficulty raising arms above the head, rising from a chair, or climbing stairs.
Balance issues and frequent falls.
Breathing problems (usually late adulthood).
Neurological examination findings:
Atrophy of tarsal muscles and tibialis anterior (usually bilaterally)
Distal weakness affecting ankle dorsiflexion, eversion, toe extension and flexion.
The weakness might progress over time to involve proximal muscles.
Muscle atrophy in the scapular region with scapular winging (A).
Waddling gait, lumbar lordosis, or scoliosis.
Decreased or absent ankle reflex
Usually intact sensation.
Usually, no facial or bulbar weakness.

Al-Tahan S et al. New family with HSPB8-associated autosomal dominant rimmed vacuolar myopathy. Neurol Genet. 2019;5:349.
Genetic Testing
Genetic testing is the primary means for diagnosis of HSPB8 Myopathy. Single-gene testing is recommended in the case of a known familial HSPB8 variant while multi-gene panel sequencing in undifferentiated cases.
Currently, HSPB8 is not included in myopathy panels in the USA. It is included in Distal Myopathy NHS panel in the UK.
HSPB8 gene can be added to diagnostic panels such as congenital myopathy panel, comprehensive myopathy panel, distal myopathy panel comprehensive Neuromuscular disorders panel, etc. by request of the ordering physician.
Providers using Invitae for genetic testing can build custom panels to include HSPB8. For details, please contact Invitae’s Client Services division, either through main phone line (1-800-436-3037) or email (clientservices@invitae.com)
Whole Exome Sequencing (WES) or Whole Genome Sequencing (WGS) can be ordered; however, the results may or may not reveal the variant. Correlation with clinical presentation, family history, other lab findings, and genetic counselling is highly recommended. Providers ordering WES and other genetic testing should include clinical notes and all required documentation to facilitate insurance coverage.
The identification of a pathogenic HSPB8 variant confirms the diagnosis in a clinically affected individual. The identification of a likely pathogenic variant may be considered diagnostic; however, further evidence would be required for reclassification of the variant.
A variant of unknown significance (VUS) is a change in HSPB8 that has not previously been associated with human disease; insufficient data exists to support whether the variant is benign or pathogenic, and therefore such a result needs further analysis as a possible new pathogenic variant before being considered as not clinically actionable. Once a diagnosis is confirmed, carrier testing is recommended for other family members.
Detailed information regarding genetic variant interpretation and pathogenicity criteria may be found by referring to American College of Medical Genetics criteria

Where to test?
* Providers using Invitae for genetic testing can build custom panels to include HSPB8. For details, please contact Invitae’s Client Services division, either through main phone line (1-800-436-3037) or email (clientservices@invitae.com)
1) NIH Genetic Testing Registry - The Genetic Testing Registry (GTR®) provides a central location for voluntary submission of genetic test information by providers. The scope includes the test's purpose, methodology, validity, evidence of the test's usefulness, and laboratory contacts and credentials. The overarching goal of the GTR is to advance the public health and research into the genetic basis of health and disease
2) Genomics England Panel App - HSPB8 gene is included in the ‘Distal Myopathies’ Panel
A crowdsourcing tool to allow gene panels to be shared, downloaded, viewed and evaluated by the Scientific Community
Other testing:
Blood tests: Elevated Creatine Kinase (CK). Some patients may have dyslipidemia and elevated liver enzymes.
Electrocardiography: Some patients might present with cardiomyopathy and conduction defects such as right bundle branch block.
Echocardiogram may show left ventricular hypertrophy and reduced ejection fraction.
Pulmonary function studies could be normal or might show respiratory insufficiency, restrictive lung disease with decreased Forced Vital Capacity (FVC)
Nerve conduction studies (NCS) showing axonal motor neuropathy predominantly affecting the lower limbs.
Electromyography (EMG) findings with features of mixed myopathic and neurogenic pathology in the upper and lower limbs.
MRI (or CT scan) of the lower limbs with fatty degenerative changes in the proximal and distal lower extremities - Figures B and C
Muscle biopsy can be done if there is diagnostic uncertainty and/or no genetic mutation is identified in genes associated with myopathies. Patients with mutations in HSPB8 gene develop myopathy displaying histologic features of myofibrillar myopathy like the pathology in myopathies due to gene defects in other components of the CASA complex such as BAG3 and DNAJB6. Muscle biopsy typically shows fatty replacement, aggregates, rimmed vacuoles, increased internalized nuclei, and endomysial fibrosis.
Management
There is no approved disease modifying therapy for HSPB8 myopathy, and the management is supportive and focuses on the quality of life.
Patients should be followed by a multidisciplinary clinic every 6 months -1 year; the components of this clinic should include a geneticist, neurologist/neuromuscular specialist, cardiologist, pulmonologist, and respiratory therapist.
Supportive care should include physical and occupational therapy, nutrition, and supplements. Other supportive measures such as mechanical aids and weight control to avoid obesity can be beneficial, and these are described in more detail in the Supportive Therapies section below.
Myopathy
HSPB8 Myopathy typically manifests in the second, third, or fourth decade of life. It is characterized by progressive muscle weakness, which may initially appear either proximally or distally. The condition predominantly affects the lower limbs and gradually spreads to the truncal muscles and upper limbs. Due to its progressive nature, individuals experience increasing difficulty with muscle strength and mobility over time.
Surveillance
Patients with HSPB8 mutations who do not currently have symptoms of myopathy, and family members who are at risk, should be screened for signs of muscle weakness via standardized strength and functional testing by a multidisciplinary team. If weakness exists, they should be followed clinically for signs of progression and possible intervention every 6 months to annually, or more frequently if progression is more rapid.
Standards of care
Unfortunately, at the moment, no formal standard of care exists for HSPB8 Myopathy. However, a standard of care has been developed and published for VCP (Valosin-containing protein) disease, thanks to the efforts of the Cure VCP advocacy group. VCP disease has many overlapping features with HSPB8 Myopathy, especially related to myopathic symptoms. Therefore, we have included the most relevant sections from the VCP standards of care below. The entire document can be accessed via this link.
Respiratory dysfunction
Clinical features
Early manifestations of respiratory involvement may include recurrent tracheobronchitis and pneumonia due to weakness of the expiratory muscles with consequent impairment of cough strength and efficacy. Progressive weakening of respiratory muscles leads to development of sleep disordered breathing characterized by nocturnal hypoventilation and oxygen desaturation. In addition, patients with bulbar symptoms are at increased risk for aspiration. Ultimately, if not properly managed, these changes can result in respiratory failure and daytime alveolar hypoventilation (i.e. hypercapnia). […]
Diagnosis
Respiratory muscle function can be assessed using pulmonary function tests [PFTs, particularly forced vital capacity (FVC): sitting and supine], maximum inspiratory and expiratory muscle pressures (MIP and MEP), and peak cough flow (PCF). Sleep disordered breathing and nocturnal hypoventilation can be identified by either a full in-lab nocturnal polysomnography or limited at-home overnight monitoring. The presence of respiratory failure and hypercapnia is often assessed using arterial blood gas analysis, but this test may not be readily available in some outpatient settings. Alternatively, measurement of the serum bicarbonate in venous blood provides an easily obtainable and reliable marker of renal compensation for chronic carbon dioxide retention. […]
Treatment
Extrapolating from management of respiratory dysfunction in other neuromuscular disorders, the treatment team should include a physical therapist, respiratory therapist, and a pulmonary specialist with expertise in management of patients with neuromuscular disease. The patient and their caregivers should maintain up to date vaccinations. Lung volume recruitment (LVR) and cough augmentation is important for the clearance of secretions and can be facilitated with LVR bag, mechanical insufflation/exsufflation (MI-E), and suction devices as needed. Respiratory infections should be treated early and aggressively. Non-invasive positive pressure ventilation (NIPPV), such as bilevel positive airway pressure (BiPAP) or volume assured pressure support (VAPS), should be initiated at the onset of nocturnal hypoventilation or with deterioration of respiratory function (e.g. arterial PCO2 >45 mmHg, FVC < 50% of predicted, MIP < − 60 cm H2O). Therapy with continuous positive airway pressure (CPAP) should be reserved for individuals that demonstrate obstructive sleep apnea in the absence of nocturnal hypoventilation. The role of respiratory muscle training to improve clinical outcomes is unclear and is currently being investigated in VCP MSP (curevcp.org).
Supplemental oxygen and/or therapy with respiratory depressants (e.g. sleep aids, narcotics, anxiolytics and antidepressants) should be withheld if possible as they lower central respiratory drive and can induce or worsen hypercapnia. Diuretic therapy may be used to reverse peripheral edema in patients with cor pulmonale. However, primary metabolic alkalosis with impaired central respiratory drive may ensue because of the associated chloride and potassium deficiency.
Surveillance
Serum bicarbonate and PFTs can be checked annually in myopathy phenotype if stable, or every 3–6 months if deteriorating […]. A sleep study should be performed at baseline in patients with documented or symptomatic weakness.
Supportive therapies (physical and occupational therapy, speech-language pathology, and respiratory therapy)
[…] Patients should have access to an individualized, comprehensive care plan with an interdisciplinary team including physical therapy (PT), occupational therapy (OT), speech language pathology (SLP), and respiratory therapy (RT). The impacts of VCP MSP across body structures and function should be routinely monitored and managed to optimize a patient’s independence with activities of daily living (ADLs) and quality of life (QOL). Clinical outcome assessments (COA) performed by trained practitioners are useful for assessing function and to track the level of independence and/or assistance required to complete ADLs. Use of standardized COAs also facilitate proactive planning for procurement of any needed assistive devices, home and work modifications, or other supports. Including a patient-reported outcome (PRO) measure with strong psychometric properties is also beneficial for assessing health indices and QOL and is recommended to be completed at least annually. Pain and fatigue PROs should also be monitored at least annually with quantitative severity scales as these may impact both physical and cognitive function. Standardized tools are preferred over generalized scales.
Physical and occupational therapy
Physical and occupational therapy focus on helping patients maintain or adapt to changes in their function and ADLs to promote independence and QOL. While surveillance by PT and OT is recommended during multidisciplinary clinic visits, if there is difficulty with performing ADLs, mobility, or transfers, patients should be referred to PT and OT for ongoing treatment including appropriate exercises, home adaptations, and/or equipment. Regular and monitored aerobic exercise programs of low to moderate intensity are recommended to preserve function, strength, range of movement, endurance, balance, independence with ADLs, and participation. Inactivity can lead to further weakness and functional decline, so maintenance of current strength versus targeted muscle strengthening with proper monitoring may be indicated in some patients. While exercise is recommended there remains a lack of evidence for the most effective prescription […]. Relevant assistive devices include ankle foot orthotics, canes, walkers, wheelchairs, chair seat risers, mobile arm supports, and hoists/ lift systems.
Speech-language pathology
A swallow study (e.g. videofluoroscopy or flexible endoscopy) can identify difficulties with swallowing permitting targeted treatment.
Respiratory therapy
RT is critical for monitoring and optimally managing respiratory function. PFTs should be performed at least annually, including measurements of FVC in sitting and supine positions. Chest physiotherapy, non-invasive ventilation, and insufflation–exsufflation devices should be initiated in a timely fashion.


Cardiomyopathy
Clinical features
The prevalence of cardiomyopathy among patients with VCP MSP is unknown but appears to be rare. VCP patients without extensive cardiac involvement may be asymptomatic, but as cardiomyopathy progresses to systolic dysfunction with increasing left ventricular dilation and pressure overload, they are more likely to become symptomatic. Patients with left ventricular failure may develop dyspnea with exercise intolerance. Physical exam findings may reveal signs of a volume overloaded state including elevated jugular venous pressure, hepatic congestion, pleural effusion with crackles, and lower extremity edema. Respiratory and cardiac failure can lead to death between the ages of 40s to 60s. A recent study showed a higher prevalence of diastolic dysfunction in VCP patients indicating it may be an early sign of cardiac involvement. It is important to exclude other etiologies of cardiomyopathy or congestive heart failure such as ischemic heart disease.
Diagnosis
Echocardiogram is useful to assess cardiac function and structure when diagnosing cardiomyopathy. However, cardiac MRI is now the gold standard for obtaining accurate cardiac dimensions and measuring function in most neuromuscular disorders because it is not hindered by anatomic abnormalities such as kyphosis.
Treatment
In patients with suspected or documented heart failure, a referral to a specialist is appropriate. Heart failure medications improve cardiac parameters in neuromuscular patients with non-ischemic cardiomyopathy. The three most useful classes of drugs for reversing cardiac remodeling related to cardiomyopathy are angiotensin-converting enzyme (ACE) inhibitors or angiotensin-receptor blockers (ARBs), beta-adrenergic receptor blockers, and mineralocorticoid receptor antagonists (MRAs). Other guideline directed medical therapies for heart failure (i.e. digoxin, diuretics, and others) have also been shown to reduce morbidity and mortality in patients with non-ischemic cardiomyopathies with a reduced ejection fraction.
If cardiomyopathy progresses despite aggressive pharmacotherapies, other treatment considerations include implantation of an automated implantable cardioverter-defibrillator (AICD) with or without biventricular pacemaker, and implantable left ventricular assist devices (LVADs).
Surveillance
Given the possibility that VCP patients with cardiomyopathy may be asymptomatic, it is reasonable to perform a cardiac MRI at baseline to determine present cardiac function. VCP patients should be monitored for symptoms of cardiomyopathy annually, and a repeat cardiac MRI can be performed if abnormal symptoms or clinical signs develop. For patients with detectable cardiac involvement on cardiac MRI such as delayed gadolinium enhancement in the ventricular wall or reduced ejection fraction, cardiac MRI should be repeated every 2 years to evaluate for cardiac remodeling following pharmacologic interventions.
Mental health
[…] Patients are at risk for higher levels of stress, depression, and anxiety due to the inherent rare nature of the disease, which predisposes them to misdiagnosis, differing clinical opinions, lack of resources and information, economic burden, and isolation. Compounding all of these issues is the absence of true disease modifying therapies for the major components of the disease […]. However, appropriate therapeutic support can help patients manage and process their diagnosis, work towards short- and long-term goals to improve their quality of life, engage in peer or professional support groups, facilitate challenging conversations with family members, reduce the risk of self-harm, and validate feelings of fear, grief, and anger.
Evidence-based practice includes screening all patients for behavioral health signs and symptoms and referring to behavioral providers upon learning of their diagnosis. Collaboration between the multidisciplinary team and community mental health providers ensures a holistic approach to supporting individuals living with VCP, both at the time of diagnosis and throughout their experience with the disease.
Useful links:
Additional Resources | Muscular Dystrophy Association (mda.org)
Mental Health Hub | Muscular Dystrophy Association (mda.org)
Supplements and nutrition
[…] It is reasonable to recommend an anti-inflammatory diet that is rich in fruits and vegetables, whole grains, lean proteins, and fatty fish, and avoid highly processed foods and products that contain preservatives, pesticides, and artificial ingredients. […] In addition, there is evidence that a Mediterranean diet may offer protection in dementia and cardiovascular disease, which could be extrapolated […], but further studies of this theory are needed. It is important to identify barriers to nutritional literacy and access to resources that may limit patients and caregivers understanding of dietary recommendations. A referral to a nutritionist or registered dietician may be appropriate.

Histological findings
HSPB8 is a chaperone involved in the Chaperone Assisted Selective Autophagy (CASA) complex. Patients with mutations in HSPB8 gene develop myopathy displaying histologic features of myofibrillar myopathy similar to the pathology in myopathies due to gene defects in other components of the CASA complex such as BAG3 and DNAJB6. Muscle biopsy typically shows fatty replacement, aggregates, rimmed vacuoles, and endomysial fibrosis.

Myopathological findings in HSPB8-related vacuolar myopathy. Biopsy of the quadriceps demonstrating multiple fibres harbouring rimmed vacuoles (arrowheads, A, haematoxylin and eosin, and B, modified Gomori trichrome) and focal increases of nicotinamide adenine dinucleotide tetrazolium reductase enzyme activity (arrowhead, C). A few fibres in one region of the specimen displayed aggregates of myotilin (arrowheads, D) and dystrophin (arrowhead, E). Rare muscle fibers showed punctate T-cell intracellular antigen 1 (TIA1) reactivity (arrowheads, F). Diffusely increased desmin, αB-crystallin and neural cell adhesion molecule reactivity was observed in few atrophic fibres (not shown). Scale bar 50 μm in all panels.
Nicolau S, Liewluck T, Elliott JL, Engel AG, Milone M. A novel heterozygous mutation in the C-terminal region of HSPB8 leads to limb-girdle rimmed vacuolar myopathy. Neuromuscul Disord. 2020 Mar;30(3):236-240. doi: 10.1016/j.nmd.2020.02.005. Epub 2020 Feb 12. PMID: 32165108.

(A) Muscle histology of a biopsy of the left vastus lateralis from case 2 shows the presence of rimmed vacuoles (in 3 fibers), adipose replacement, moderate to severe endomysial fibrosis, muscle fiber size variation, numerous atrophic muscle fibers, and increase in central nuclei. (B) Semithin section stained with toluidine blue showing a single fiber with accumulated autophagic vacuoles, corresponding to rimmed vacuolar changes in muscle cryosections.
Al-Tahan S, Weiss L, Yu H, Tang S, Saporta M, Vihola A, Mozaffar T, Udd B, Kimonis V. New family with HSPB8-associated autosomal dominant rimmed vacuolar myopathy. Neurol Genet. 2019 Jul 10;5(4):e349. doi: 10.1212/NXG.0000000000000349. PMID: 31403083; PMCID: PMC6659134.
Muscle biopsy shows rimmed vacuoles:
Learn More
About HSPB8 Myopathy

We invite clinicians to expand their knowledge on HSPB8 Myopathy by watching a video on Vumedi, a global video education platform for doctors worldwide.
In this video, Dr. Lan Weiss from UCI provides an introduction to HSPB8 Myopathy and discusses the latest preclinical translational research. To access the video, please click on the image below.

Disease Classification
Orphanet, a database of rare diseases, and National Organization for Rare Diseases (NORD) classify HSPB8 Myopathy as a subtype of Myofibrillar Myopathy (MFM). Some sources describe it as a form of LGMD. Below, Dr. Alyaa Shmara discusses why we believe HSPB8 Myopathy falls under these categories:
HSPB8 Myopathy is also known as
HSPB8 Inclusion Body Myopathy with Rimmed Vacuoles
HSPB8 Rimmed Vacuolar Myofibrillar Myopathy (Ghaoui et al., 2016)
Autosomal dominant distal axonal motor neuropathy-myofibrillar myopathy syndrome (Orphanet)
Limb-girdle rimmed vacuolar myopathy (Nicolau et al., 2020)
Multisystem proteinopathy (MSP) or MSP-like disorder (Chompoopong et al., 2024)
Autosomal dominant rimmed vacuolar myopathy (Al Tahan et al., 2019)
ORPHA:476093
HSPB8 as MFM
Myofibrillar myopathies are a group of rare genetic neuromuscular disorders that may be diagnosed in childhood but most often appear after 40 years of age. These conditions are highly variable but are characterized by a slowly progressive muscle weakness that can involve skeletal muscle (muscles that function to move bones) and smooth muscle (muscle often associated with organs, such as the digestive tract). Skeletal muscle weakness can be present in the limb muscles close to the center of the body (proximal) as well as the muscle farther from the center of the body (distal). A weakening of the heart muscle (cardiomyopathy) is common and may result in an irregular heartbeat (arrhythmia or conduction defects) or congestive heart failure.
HSPB8 is part of the chaperone-assisted selective autophagy (CASA) complex, a vital part of the cellular protein quality control system in mechanically strained cells and tissues such as skeletal muscle, heart, and lung. In muscle, CASA maintains the Z-disk and protein turnover. CASA mediates degradation of the actin cross-linking protein filamin. If mechanical tension results in permanent unfolding of filamin, it is recognized by the CASA chaperone complex and leads to autophagic degradation of damaged proteins [1].
Mutations in HSPB8 gene, which have previously been associated with Charcot-Marie-Tooth type 2L and distal hereditary motor neuronopathy type IIa, have also been reported to associate with autosomal dominant Rimmed Vacuolar Myopathy. Affected patients have distal and proximal limb girdle myopathy. Muscle biopsy displays histologic features of myofibrillar myopathy with aggregates and rimmed vacuoles.
Ghaoui et al identified 2 families with distal weakness progressing to involve proximal muscles with mutations in HSPB8 causing a dual involvement of a peripheral motor neuropathy and a rimmed vacuolar myofibrillar myopathy. Muscle biopsy shows evidence of a myofibrillar autophagic myopathy, rimmed vacuoles, and protein aggregates positive for proteins associated with myofibrillar myopathy such as desmin, myotilin, and α-B-crystallin. These aggregates also contain HSPB8 and other CASA complex partners DNAJB6 and BAG3 [3,4] (Fig 1.) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4776089/


Laguna et al. identified three families with a HSPB8 myopathy. Muscle biopsies showed dystrophic features with atrophy, necrosis and regeneration, internal nuclei, fibrosis, fiber splitting and rimmed vacuoles. No neurogenic aspect was seen. Protein aggregations were revealed using ubiquitin and desmin antibodies (Fig.2: 4–5). TDP-43 antibody revealed coarse cytoplasmic aggregation and linear sarcolemmal expression (Fig.2: 6). EM studies showed focal disruption of the myofibrillar network, Z lines streaming and areas lacking myofibrils containing granulofilamentous material (Fig.2: 7–8). HSPB8 haploinsufficiency causes dominant adult-onset axial and distal myopathy | Acta Neuropathologica (springer.com)
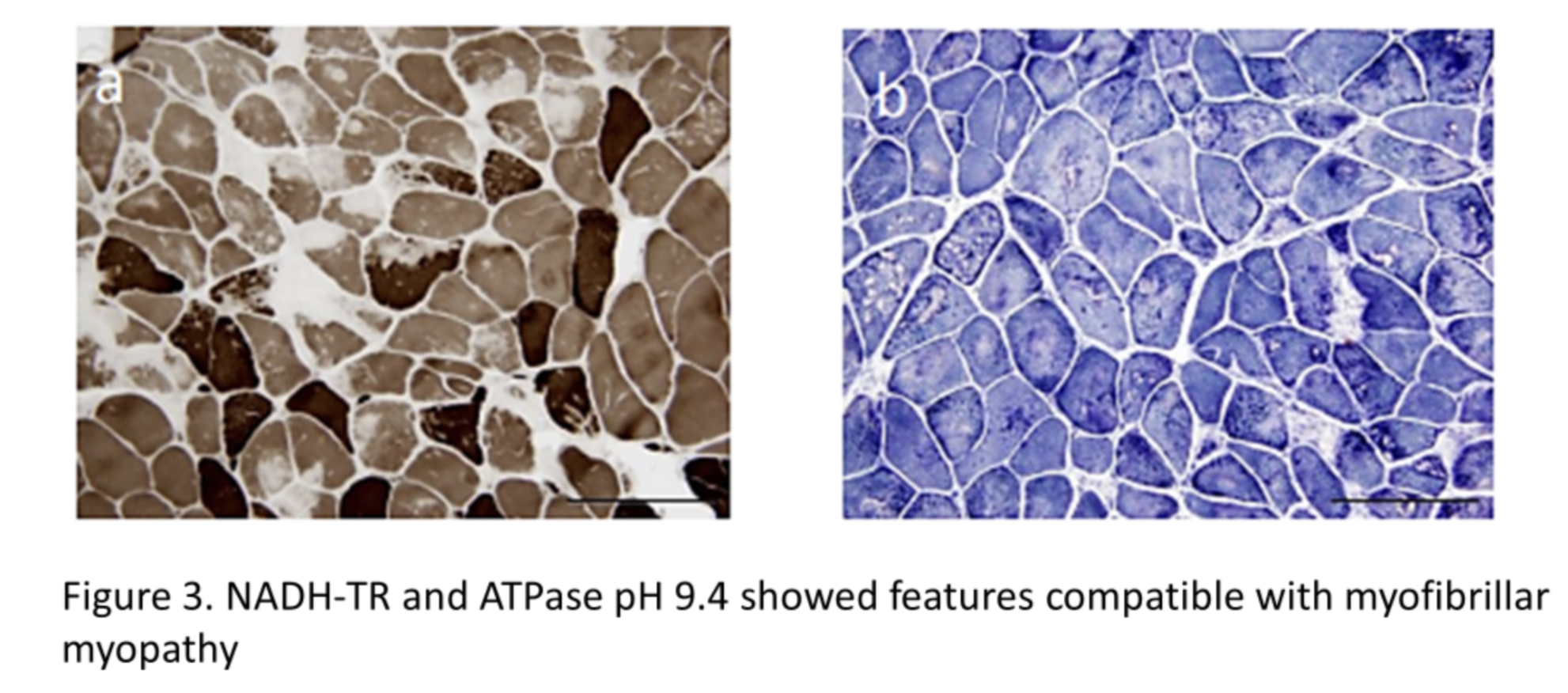
NADH-TR and ATPase pH 9.4 showed features compatible with myofibrillar myopathy [4] (Fig. 3) 401_2017_1724_MOESM1_ESM.pdf (springer.com)
References:
Arndt V, Dick N, Tawo R, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol. 2010;20(2):143-148. doi:10.1016/j.cub.2009.11.022
Sarparanta J, Jonson PH, Golzio C, et al. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nature Genetics. 2012 Feb;44(4):450-5, S1-2. DOI: 10.1038/ng.1103. PMID: 22366786; PMCID: PMC3315599.
Ghaoui, Roula et al. “Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy.” Neurology vol. 86,4 (2016): 391-8. doi:10.1212/WNL.0000000000002324
Echaniz-Laguna, A., Lornage, X., Lannes, B. et al. HSPB8 haploinsufficiency causes dominant adult-onset axial and distal myopathy. Acta Neuropathol 134, 163–165 (2017). https://doi.org/10.1007/s00401-017-1724-8
Section Author : Dr Alyaa Shmara